
![[CV1100] GetClone™ 零背景平末端載體選殖載體, 20 RXN](/web/image/product.template/347/image?unique=087243d)
![[CV1100] GetClone™ 零背景平末端載體選殖載體, 20 RXN](/website/image/product.template/347/image/90x90)

Description
The GetClone™ PCR Cloning Vector II is a positive selection system for high efficiency cloning of blunt end DNA or amplicons. This cloning vector contains a lethal gene which can be disrupted by ligation of a blunt end DNA insert into the cloning site. Only colonies with inserted vectors are able to propagate, eliminating the additional needs of IPTG and X-Gal for blue/white screening. This cloning vector includes ampicillin and kanamycin resistance genes that can meet the needs of most users.
Features
Cloning efficiency greater than 90%
IPTG and X-Gal are not required
Accepts a wide range of insert/vector ratios 0.5:1 to 12:1
Accepts insert size from 6 bp to 11 kb
The phosphorylation of PCR fragments is not required
Accepts blunt end amplicon or DNA fragment (not for sticky ends)
Ampicillin and kanamycin selection markers
Storage
-20°C for 24 months
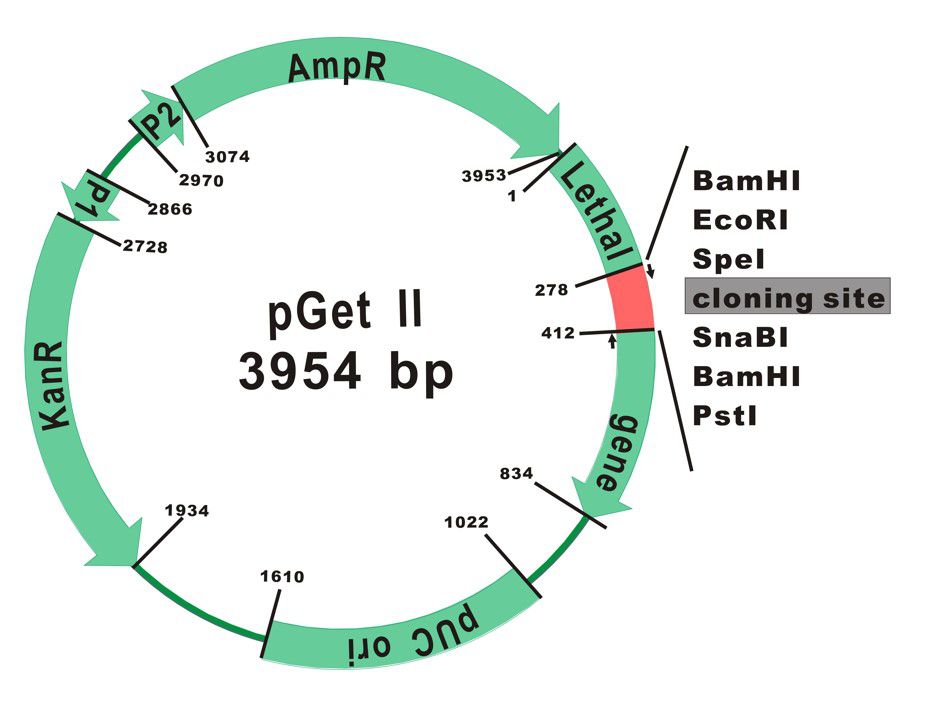
The plasmid map of pGet II Vector
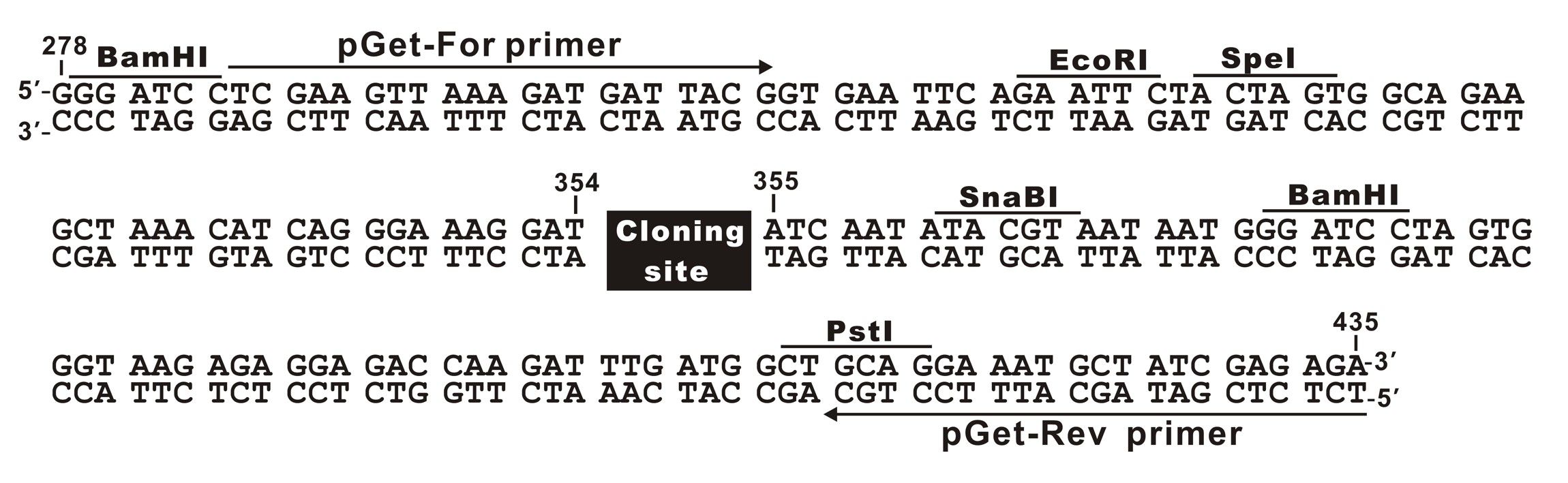
The plasmid cloning sites of pGet II Vector
Contents
Component | Volume |
pGet II Vector (25 ng/μl) | 23 μl |
pGet-For Primer (10 μM) | 100 μl |
pGet-Rev Primer (10 μM) | 100 μl |
Primers Sequence
pGet-For Primer:
5'-TCGAAGTTAAAGATGATTACGG-3'
pGet-Rev Primer:
5'-TCTCTCGATAGCATTTCCTGC-3'
Storage
-20°C for 24 months
Manual
Manual_CV1100_GetClone™ PCR Cloning Vector II (2023 ver 2.3.0)
Sequence in Fasta format
Sequence_CV1100_GetClone™ PCR Cloning Vector II
SDS
Why use the GetClone™ Vector for PCR cloning instead of a T-vector?
For conducting PCR-cloning, a high fidelity polymerase is required to keep accuracy of PCR amplification. In general, high fidelity enzymes vetting correctness such as SMO-HiFi™, Pfu, KOD, and Phusion are associated with proofreading activity which results in blunt ends of PCR products. GetClone™ Vector can directly accept blunt-ended PCR fragments for ligation, without the need of extra step of DNA phosphorylation. As contrast, T-vector used for TA-cloning requires DNA fragment with an extra Adenosine nucleotide added at the 3’ end which is usually generated by non-proofreading and thus less fidelity DNA polymerase like Taq polymerase.
Is blunt end ligation more difficult than sticky end ligation?
It depends on the length of sticky end. Generally, sticky end is easier to perform in comparison to blunt end ligation. However, a sticky end with only one extruding base as seen in the TA-cloning is probably more difficult for ligation as compared with a blunt end (also referred to NEB for NciI or EcoNI instructions).
After using GetClone™ Vector for PCR cloning, how can the colonies carrying the cloned fragment be selected?
In most cases of using GetClone™ Vector, colonies grown on a plate with appropriate antibiotics should be the positive clones because a bacterium accepting an empty GetClone™ Vector without an insert would fail to proliferate. To further identify the clones, it is recommended to use Colony-PCR method. In brief, the colony grown on the plate is directly picked up with a toothpick and then mixed into a PCR premix (ex.: SMOBIO TP1200 or TP1260) and PCR amplification is conducted in the thermal cycler with primers (pGET-For and pGET-Rev) which are also provided with GetClone™ Vector. The expected length of PCR product should be the one of the insert plus 152 bp.
How do I perform sequencing when using the GetClone™ Vector?
Please use the GetClone™ Vector primers for sequence service.
In use of GetClone™ Vector for cloning, why are some colonies in absence of inserts?
Several reasons can be stated as follows:
Reason 1: Check your insert. If it is not blunt-end, it cannot be ligated with GetClone™ vector, as this may result in false positive results for majority of the colonies.
Reason 2: When DNA concentration of the insert is too low, the successful ligates are largely reduced in proportion, leading to an overwhelming vector background. The solution is to increase the concentration of the insert DNA.
Reason 3: If the target DNA fragments for cloning are large, such as 10kb or more, transformation efficiency depreciates after ligation. This results in fewer colonies having insertions. Therefore, it is suggested to increase the insert DNA concentration and reduce the concentration of vector DNA to re-execute ligation. It will also enhance chances to get the right clone by using higher efficiency competent cells.
Reason 4: Non-specific PCR products or/and primer dimers which might occurred during PCR amplification are supposed to compete or interfere the ligation of target PCR fragment with the vector, and thus reducing the number of correct clones. Before conducting ligation, it is therefore recommended to isolate the desired DNA fragment from other non-specific DNA by gel extraction.
If I did not add any insert DNA fragment for ligation with GetClone™ Vector, why are colonies being grown on the selective plate?
This is probably due to generation of mutation on the vector DNA during cloning. A mutated lethal gene can lead to false positive colonies, despite that the occurrence is rare.
How to count the 90% cloning efficiency of GetClone™?
The GFP report gene was PCR amplified as the insert to the GetClone™ Vector. After ligation and transformation, positive clones (with insert) presenting green fluorescence on the plate were calculated in number, and ~ 90% of total colonies did shine as observed in B-Box, demonstrating the high cloning efficiency of GetClone™ Vector .
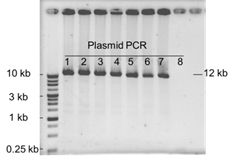
Do you have any experiment data for cloning 12 kb fragment with GetClone™ Vector?
Yes, we do have the data as shown below. Eight colonies were picked up for plasmid isolation and PCR analysis. It was observed that seven out of eight colonies (87.5%) were positive clones which could be verified by successful amplification of 12.1 kb fragments.
Ligation Example 1 (NEB T4 DNA Ligase #M0202)
Insert (Blunt end) X μl (Y ng*)
pGet II (3954 bp) 1 μl (25 ng)
Mix well then add
10X T4 DNA Ligase Buffer 2 μl
T4 DNA Ligase 1 μl
ddH2O to 20 μl
Final volume 20 μl
Mix well then incubate at 16°C or room temperature (20~25°C) for 1 hours.
Ligation Example 2 (TOYOBO Ligation High ver2 #LGK-201)
Insert (Blunt end) X μl (Y ng*)
pGet II (3954 bp) 1 μl (25 ng)
ddH2O up to 7 μl
Ligation high ver2 3.5 μl
Final volume 10.5 μl
Mix well then incubate at 16°C or room temperature (20~25°C) for 5~30 mins.
*For 3/1 of Insert/Vector molar ratio:

Transformation
The GetClone™ is compatible with most available competent E. coli cells. Apply 1 ~10 µl of ligation mixture to 10 times volume competent E. coli cells. Perform transformation procedures according to the instruction of the competent cells. Spread the transformed E. coli cells on an LB-ampicillin (50~100 µg/ml) or LB-Kanamycin (50 µg/ml) plate for colony selection.
Recommended colony PCR condition
(SMOBIO’s TP1200 ExcelTaq™ 5X PCR Master Dye Mix is suggested)
Template | Single colony |
pGet-For Primer | 0.5 µl |
pGet-Rev Primer | 0.5 µl |
5X PCR Master Mix | 5 µl |
H2O | to 25 µl |
Total volume | 25 µl |
Recommended PCR Program
Steps | Temp. | Time | Cycles |
Template denature | 94°C | 2 min | 1 |
Denature | 94°C | 30 sec | 25-40 |
Annealing | 50°C | 30 sec | |
Extension | 72°C | 30 sec/kb | |
Final extension | 72°C | 1 min | 1 |
PMCID: PMC9130408
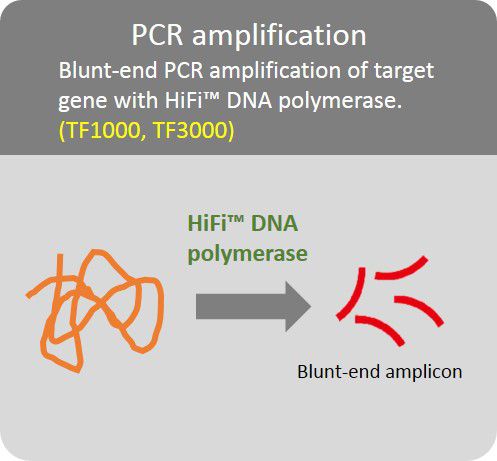
High Fidelity PCR amplification
Amplification of target gene with HiFi™ DNA polymerase to minimize error rate.

Gel electrophoresis
Staining amplicons with safe fluorescent dyes, following by observation under blue-light illuminator to minimize damage of DNA amplicons and maximize successful cloning efficiency.
Safe fluorescent dyes
[NS1000] FluoroVue™ Nucleic Acid Gel Stain (10,000X), 500 μl
[DS1000] FluoroStain™ DNA Fluorescent Staining Dye (Green, 10,000X), 500 μl
[DL5000] FluoroDye™ DNA Fluorescent Loading Dye (Green, 6X), 1 ml
Blue-light illuminator
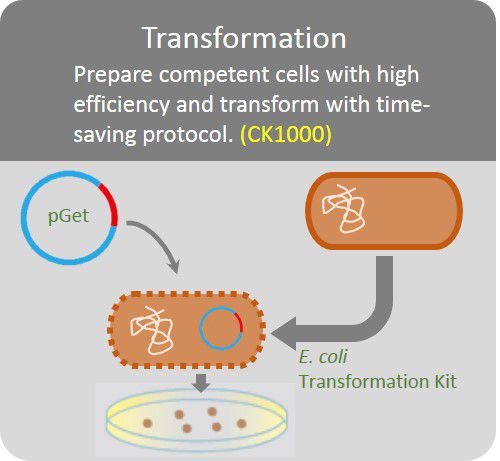
Transformation
Prepare competent cells with high efficiency and transform with time-saving protocol.
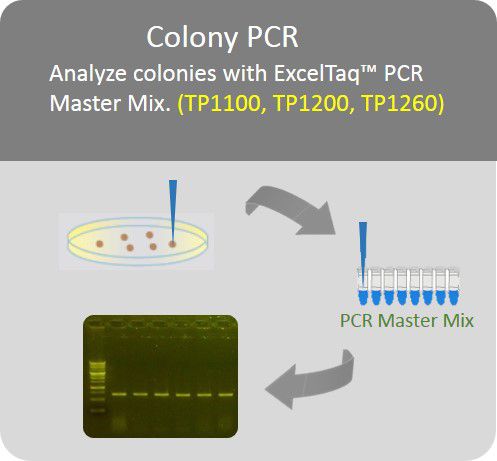
Colony PCR
Analyze colonies with PCR master mix to save preparation time.